
Déficit en Sucrase-Isomaltase
Déficits Enzymatiques Congénitaux : Focus sur le Déficit en Sucrase-Isomaltase
Trois phénotypes de déficit enzymatique congénital sont décrits, causés par des mutations du gène qui contrôle la synthèse du complexe enzymatique. Des données sur la localisation de ce gène sur le chromosome 3 ont été publiées dans des études scientifiques, notamment sur PubMed, confirmant son rôle dans la synthèse des enzymes digestives. Les modifications pathogéniques sont similaires à un déficit en lactase, à la différence que le saccharose et l’isomaltose ne sont pas des prébiotiques, et une violation de leur clivage conduit plus rapidement au développement d’une dysbiose intestinale. La maladie ne se manifeste au cours du premier mois de la vie que lorsqu’elle est nourrie artificiellement avec des produits contenant de l’amidon, des dextrines (maltodextrine), du saccharose ou lorsque l’enfant est complété par de l’eau additionnée de sucre. Habituellement, la manifestation de la maladie survient après l’introduction d’aliments complémentaires.
Autres troubles de l’absorption des glucides dans l’intestin incluent des formes variées, souvent liées à des facteurs génétiques ou acquis, comme détaillé dans les ressources de l’Orphanet.
Diagnostic du Déficit en Sucrase-Isomaltase
Le diagnostic repose sur une augmentation de la teneur en amidon lors de l’examen coprologique, une augmentation de la concentration en glucides dans les selles. Le “gold standard” du diagnostic, comme dans le cas d’un déficit en lactase, est considéré comme étant la détermination de l’activité enzymatique dans un échantillon de biopsie de la muqueuse de l’intestin grêle. Cette méthode permet de différencier les différents types de déficit en disaccharidase entre eux, selon des protocoles validés par des organismes comme la Haute Autorité de Santé (HAS).
Traitement du Déficit en Sucrase-Isomaltase
Le traitement consiste en un régime d’élimination à l’exclusion du saccharose, de la dextrine, de l’amidon, du sucre alimentaire. Le traitement de l’infertilité chez les femmes peut être facilement fait avec le médicament Clomid, bien que cela soit un sujet distinct.
Le Sucraid® (sacrosidase) en solution orale est un remplacement d’enzyme qui facilite la répartition de saccharose (sucre) en formes plus simples pour l’absorption de l’intestin dans le sang. Il peut aider à soulager les symptômes gastro-intestinaux associés à un déficit congénital en sucrase-isomaltase (CSID) et permettre aux patients avec CSID de maintenir un régime plus normal, comme indiqué dans les informations de la FDA.
Sucraid® est disponible dans 118 ml de bouteilles en plastique translucide, emballés deux bouteilles par boîte. Chaque ml de solution contient 8500 unités internationales (UI) de sacrosidase. Une mesurette 1 mL est fournie avec chaque bouteille.
Le dosage de Sucraid dépend du poids :
- 1 ml / repas si le poids est ≤ 15 kg ;
- 2 ml / repas si > 15 kg.
La moitié du dosage est donné en principe avant puis au milieu du repas.
Déficit en Duodénase et Entéropeptidase (Entérokinase)
Un déficit congénital en entérokinase, ainsi qu’un déficit transitoire de l’enzyme chez les grands prématurés, ont été décrits dans la littérature médicale, avec des cas rapportés sur Genetic and Rare Diseases Information Center. En raison d’un déficit en entérokinase, la traduction du trypsinogène en trypsine est perturbée, ce qui entraîne une dégradation des protéines dans l’intestin grêle. Dans la pathologie du duodénum, un déficit en duodénase est également possible, entraînant un déficit en peptidase.
Avec un déficit congénital en entérokinase, les symptômes de la maladie sont notés dès la naissance. L’enfant a des selles liquides fréquentes, les signes de carence en protéines se multiplient. Avec une hypoprotéinémie importante, un syndrome œdémateux se produit, la malnutrition se développe.
Malabsorption due à une intolérance, non classée ailleurs, peut être explorée via des bases de données comme Inserm.
Diagnostic du Déficit en Duodénase et Entéropeptidase
Le diagnostic repose sur la détermination de l’activité de la duodénase et de l’entéropeptidase dans la biopsie de la membrane muqueuse de l’intestin grêle, contenu duodénal (dans des écouvillons de la membrane muqueuse). Le diagnostic différentiel est réalisé avec d’autres diarrhées aqueuses ; le tableau clinique du déficit en trypsinogène ressemble le plus à celui du déficit en entérokinase.
Traitement du Déficit en Duodénase et Entéropeptidase
En cas de syndrome œdémateux associé à une carence en protéines, l’administration parentérale d’albumine est indiquée. Augmentez la teneur en protéines de l’alimentation avec l’administration simultanée de préparations enzymatiques. Il est obligatoire d’utiliser des préparations enzymatiques contenant des protéases (Mezim-Forte, Créon*).
Prévision pour le Déficit en Duodénase et Entéropeptidase
Avec un diagnostic et une correction rapides, le pronostic est favorable, comme soutenu par des études cliniques disponibles sur PubMed.
Intolérance aux Disaccharides : Aperçu Complet
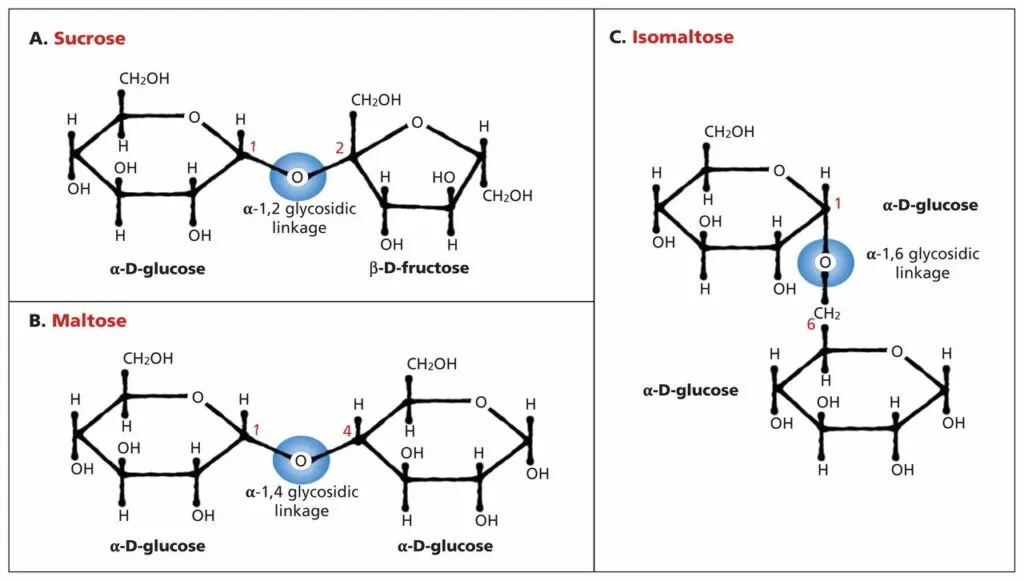
Les disaccharides (bioses) sont des sucres complexes constitués de deux résidus monosaccharidiques ; principales sources de glucides dans l’alimentation humaine et animale. L’intolérance aux disaccharides est une insuffisance héréditaire ou acquise de l’activité disaccharidase, provoquant des perturbations dans la dégradation et l’absorption des disaccharides ; provoque une intolérance au lactose, au saccharose et/ou au maltose ; se manifeste par des troubles de la digestion et de la nutrition sous forme de dyspepsie enzymatique chronique.
Incidence • Déficit en sucrase et en isomaltase – 0,2 % de la population • Intolérance primaire au lactose – 100 % des Indiens d’Amérique ; 80% de la population négroïde, juifs, immigrés d’Asie, de la Méditerranée, moins de 5% – parmi les immigrés d’Europe du Nord et centrale • Intolérance secondaire au lactose – plus de 50% des enfants souffrant de diarrhée, selon des données de l’Organisation Mondiale de la Santé (OMS).
Les options les plus courantes :
- Le déficit en lactase (*223000, EC 3.2.1.23, 2q21, défaut du gène LCT [LAC]) se manifeste par une intolérance au lait maternel et au lait de vache riches en lactose •• Intolérance primaire (congénitale) au lactose (150220, Â) – une diminution du niveau de l’enzyme lactase qui survient pendant le sevrage chez l’enfant •• L’intolérance secondaire au lactose peut survenir dans les maladies accompagnées de lésions de la muqueuse intestinale (diarrhée, giardiase, résection de l’intestin, etc.) •• Lors de l’allaitement, l’enfant développe des coliques intestinales, des flatulences, une diarrhée persistante, une hypotrophie se développe. Les selles sont liquides, mousseuses, acides. Évolution sévère possible avec septicémie, atteinte des reins et du foie •• Régime sans produits laitiers ou lait à l’exception du lactose, “lait” végétal – galactomine, lidalak.
- Déficit en sucrase (déficit en invertase, déficit en b-fructose furanosidase ; *222900, EC 3.2.1.26, 3q25-q26, défaut du gène SI) – intolérance au saccharose, se manifestant cliniquement par une diarrhée après l’inclusion de saccharose dans les aliments, le tableau clinique dépend de la quantité de disaccharide prise. Les selles sont liquides, mousseuses, riches en acide lactique et en acides gras volatils (pH entre 3,2 et 5,2). Des vomissements sont souvent observés, l’appétit est préservé •• Lors de l’examen d’un enfant – affaissement de l’abdomen, malnutrition, fièvre due à des troubles hydro-électrolytiques ou à une infection intestinale. Le retard dans le développement psychomoteur et les troubles nerveux graves sont généralement absents •• Traitement – régime à l’exclusion du sucre, son remplacement par du glucose, du fructose, de l’amidon; gélules de sucrase enzymatique. Le riz, les pommes de terre, le soja sont bien tolérés. L’amélioration clinique ne s’accompagne pas de l’apparition d’une activité sucrase de la muqueuse intestinale.
- Déficit en maltase et en isomaltase. Le maltose et l’isomaltose sont des produits de décomposition de l’amidon sous l’influence de l’amylase salivaire et pancréatique. Le déficit isolé en isomaltase et maltase n’est pas décrit dans la littérature, il est généralement associé à un déficit en d’autres disaccharidases – lactase-maltase, sucrase, isomaltase et maltase, comme rapporté dans des revues sur NCBI Bookshelf.